
(一)技术领域
本发明涉及一种脱除羟基的苄基类保护基的方法。
(二)背景技术
羟基是有机合成中一个重要的官能团,通过反应其可转变成为多种其它的官能团。由于羟基比较活泼,所以在多步有机合成反应中经常需要使用保护基将其保护起来,然后在适当的时候脱去保护基。在各种羟基保护基团中,苄基类保护基,包括苄基以及取代苄基,对于各种试剂和反应条件的稳定性较好,是应用最广泛的保护基之一。因此脱除羟基的苄基类保护基团在有机合成中也尤为重要。
脱除羟基的苄基类保护基团的方法有很多种,其中一种是以2,3-二氯-5,6-二腈基-1,4-苯醌(DDQ)为氧化剂进行氧化脱保护。例如,天然产物(+)-Laurencin的全合成过程中使用DDQ脱除了羟基的苄基保护基(Org.Lett.2005,7:75-77);文献(Tetrahedron 1986,42:3021-3028)报道了DDQ可有效脱除羟基的4-甲氧基苄基保护基和3,4-二甲氧基苄基保护基;诺华公司目前处于临床前测试的抗癌化合物Discodermolide生产过程中两次使用DDQ氧化脱除羟基的4-甲氧基苄基保护基(Chem.Rev.2006,106:2943-2989)。在使用DDQ脱除羟基的苄基类保护基的反应中,DDQ作为化学氧化剂的用量一般是化学计量或超过化学计量的,这降低了反应的经济性,并且增加了产物分离纯化的难度。为了克服以上缺点,文献(Tetrahedron Lett.2000,41:10323-10326和Tetrahedron Lett.2001,42:5571-5573)以DDQ为催化剂,以底物三倍计量的Mn(OAc)3为氧化剂进行氧化脱保护分别脱除了羟基的4-甲氧基苄基保护基和4-苯基苄基保护基;文献(Org.Lett.2010,12:4686-4689)以DDQ为催化剂,以底物六倍计量的Mn02为氧化剂进行氧化脱保护脱除了羟基的4-甲氧基苄基保护基。这些方法虽然降低了DDQ的用量,但不可避免产生大量的固体废弃物或含金属盐的废水,对环境造成不利影响。
(三)发明内容
本发明要解决的技术问题是提供一种经济、环保的除去羟基的苄基类保护基的方法。
为解决上述技术问题,本发明采用如下技术方案:
一种脱除羟基的苄基类保护基的方法,所述的方法为:以羟基化合物的羟基上苄基类保护基得到的带有苄基类保护基的化合物为反应底物,以2,3-二氯-5,6-二腈基-1,4-苯醌(DDQ)和亚硝酸叔丁酯(TBN)为催化剂,以氧气为氧化剂,反应底物在有机溶剂中,于氧气压力不大于0.5MPa、温度100~140℃的条件下进行反应,脱除羟基的苄基类保护基得到羟基化合物。
本发明所述苄基类保护基可以是苄基、4-甲氧基苄基、2-甲基苄基、3-甲基苄基、4-甲基苄基或3,4-二甲氧基苄基,优选4-甲氧基苄基。
本发明中,所述的羟基化合物为脂肪醇(例如C1~C20的脂肪醇)、脂环醇(例如3~8元环结构的脂环醇)、饱和或不饱和杂环醇或带有其它官能团的醇,并且可以为一元醇或多元醇。所述的饱和或不饱和杂环醇可以是含N、O等杂原子的饱和或不饱和杂环醇,例如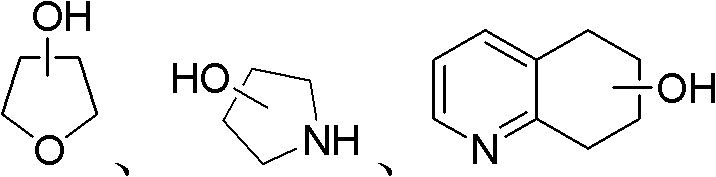 等。所述的带有其他官能团的醇可以是取代的脂肪醇、取代的脂环醇或者取代的饱和或不饱和杂环醇,所述的取代的脂肪醇是指脂肪醇的烃基被一个或多个取代基取代,所述的取代基各自独立选自下列之一:卤素、C1-C4的烷氧基、酯基、苯基、饱和杂环基团,所述的饱和杂环基团可以是含N、O等杂原子的饱和杂环基团,如
等。所述的带有其他官能团的醇可以是取代的脂肪醇、取代的脂环醇或者取代的饱和或不饱和杂环醇,所述的取代的脂肪醇是指脂肪醇的烃基被一个或多个取代基取代,所述的取代基各自独立选自下列之一:卤素、C1-C4的烷氧基、酯基、苯基、饱和杂环基团,所述的饱和杂环基团可以是含N、O等杂原子的饱和杂环基团,如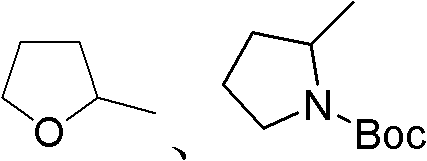 等;所述的取代的脂环醇是指脂环醇的环上被一个或多个取代基取代,所述的取代基各自独立选自下列之一:C1-C4的烷基、卤素、C1-C4的烷氧基、酯基、叔丁氧羰基;所述的取代的饱和或不饱和杂环醇是指饱和或不饱和杂环醇的杂环上被一个或多个取代基取代,所述的取代基各自独立选自下列之一:C1-C4的烷基、卤素、C1-C4的烷氧基、酯基、叔丁氧羰基、缩醛基。
等;所述的取代的脂环醇是指脂环醇的环上被一个或多个取代基取代,所述的取代基各自独立选自下列之一:C1-C4的烷基、卤素、C1-C4的烷氧基、酯基、叔丁氧羰基;所述的取代的饱和或不饱和杂环醇是指饱和或不饱和杂环醇的杂环上被一个或多个取代基取代,所述的取代基各自独立选自下列之一:C1-C4的烷基、卤素、C1-C4的烷氧基、酯基、叔丁氧羰基、缩醛基。
进一步,所述的羟基化合物优选下列之一:正辛醇、2-辛醇、苯乙醇、环己醇、甲基环己醇、己二醇、N-Boc-L-脯氨醇、L-薄荷醇、四氢糠醇、2-氯-5,6,7,8-四氢-8-羟基喹啉、双丙酮葡萄糖、6-甲氧基-1-己醇、6-(甲氧基甲氧基)-1-己醇、1,6-己二醇单乙酸酯或1,6-己二醇单苯甲酸酯。
本发明所述有机溶剂优选邻二氯苯、间二氯苯或乙二醇二乙醚,优选乙二醇二乙醚。
本发明所述催化剂DDQ与反应底物的投料摩尔比为3~15∶100,优选5~10∶100。
本发明所述催化剂TBN与反应底物的投料摩尔比为3~15∶100,优选5~10∶100。
本发明中,所述有机溶剂的质量用量推荐为反应底物的3~15倍。
本发明中,所述氧气的压力优选为0.1~0.5MPa,更优选0.2~0.3MPa。
本发明中,所述反应温度为100~140℃,优选为120~125℃。
本发明中,所述反应时间通常为30~240min,优选为50~120min。
本发明具体推荐所述脱除羟基的苄基类保护基的方法按照以下步骤进行:将反应底物溶于有机溶剂中,加入2,3-二氯-5,6-二腈基-1,4-苯醌和亚硝酸叔丁酯,以0.2~0.3MPa的氧气为氧化剂,在120~125℃下反应50~120min,脱除苄基类保护基,得到羟基化合物;所述的有机溶剂是邻二氯苯、间二氯苯或乙二醇二乙醚;所述的2,3-二氯-5,6-二腈基-1,4-苯醌、亚硝酸叔丁酯与反应底物的投料摩尔比为5~10∶5~10∶100。
在上述脱除苄基类保护基的反应完全后,可以采用常规过柱分离得到羟基化合物。
本发明操作简便安全,其有益效果主要在于:
A)与传统的使用化学计量的DDQ氧化脱保护法相比,本发明中DDQ的用量大大减少,降低了反应成本。
B)与以DDQ为催化剂、金属氧化物或金属盐为氧化剂的氧化脱保护法相比,本发明中使用了清洁的氧气为氧化剂,大大降低了环境成本。
(四)具体实施方式
下面通过具体实施方式对本发明作进一步说明,但本发明的保护范围并不限于此。
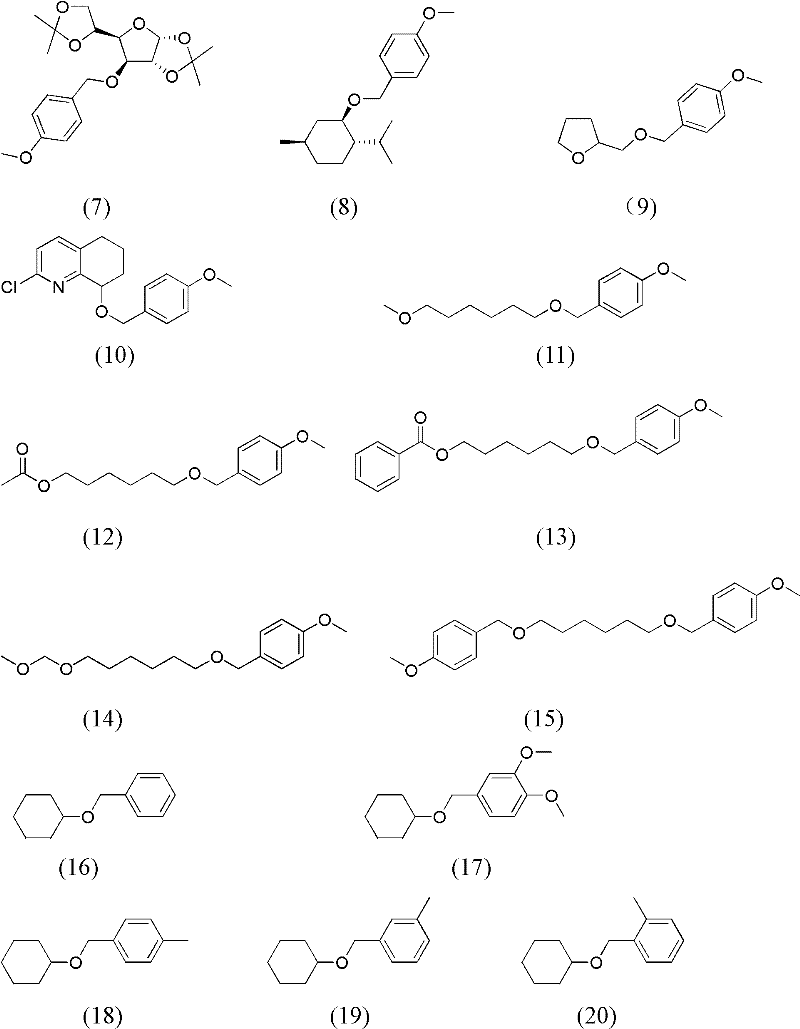
下述实施例所用的带有苄基类保护基的反应底物的结构式分别如式(1)~(20)所示:
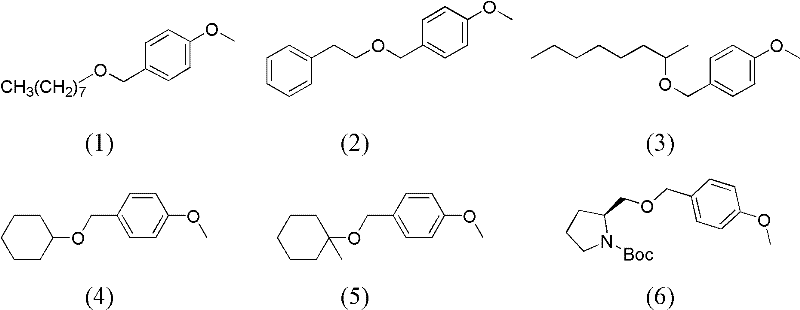
实施例1:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的正辛醇(式(1)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应120min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,正辛醇的分离收率为93%。
实施例2:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的苯乙醇(式(2)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应120min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,苯乙醇的分离收率为95%。
实施例3:
反应步骤同实施例2,所不同的是乙二醇二乙醚改为邻二氯苯,转化率为100%,产物选择性为99%。苯乙醇的分离收率为95%。
实施例4:
反应步骤同实施例2,所不同的是充氧气至压力表为0.3MPa,反应时间为105min,转化率为98%,产物选择性为99%。苯乙醇的分离收率为94%。
实施例5:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的2-辛醇(式(3)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应90min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,2-辛醇的分离收率为94%。
实施例6:
反应步骤同实施例5,所不同的是DDQ用量为0.24mmol,反应时间为160min,转化率为100%,产物选择性为98%。2-辛醇的分离收率为93%。
实施例7:
反应步骤同实施例5,所不同的是TBN用量为0.24mmol,反应时间为155min,转化率为98%,产物选择性为98%。2-辛醇的分离收率为92%。
实施例8:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的环己醇(式(4)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应90min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,环己醇的分离收率为93%。
实施例9:
反应步骤同实施例8,所不同的是乙二醇二乙醚改为间二氯苯,转化率为100%,产物选择性为99%。环己醇的分离收率为93%。
实施例10:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的1-甲基环己醇(式(5)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应90min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,1-甲基环己醇的分离收率为94%。
实施例11:
反应步骤同实施例10,所不同的是充氧气至压力表为0.5MPa,反应时间为60min,转化率为100%,产物选择性为98%。1-甲基环己醇的分离收率为94%。
实施例12:
反应步骤同实施例10,所不同的是充氧气至压力表为0.1MPa,反应时间为140min,转化率为97%,产物选择性为99%。1-甲基环己醇的分离收率为92%。
实施例13:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的N-Boc-L-脯氨醇(式(6)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应105min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,N-Boc-L-脯氨醇的分离收率为93%。
实施例14:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的双丙酮葡萄糖(式(7)),10mL乙二醇二乙醚,0.96mmol的DDQ,0.96mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应35min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为91%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,双丙酮葡萄糖的分离收率为84%。
实施例15:
反应步骤同实施例14,所不同的是DDQ用量为1.2mmol,TBN的用量为1.2mmol,反应时间为30min,转化率为100%,产物选择性为95%。双丙酮葡萄糖的分离收率为89%。
实施例16:
反应步骤同实施例14,所不同的是乙二醇二乙醚的用量为50mL,转化率为98%,产物选择性为91%。双丙酮葡萄糖的分离收率为83%。
实施例17:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的L-薄荷醇(式(8)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应105min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,L-薄荷醇的分离收率为95%。
实施例18:
反应步骤同实施例17,所不同的是油浴的温度为125℃,反应时间为95min,转化率为100%,产物选择性为99%。L-薄荷醇的分离收率为95%。
实施例19:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的四氢糠醇(式(9)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应100min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,四氢糠醇的分离收率为94%。
实施例20:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的2-氯-5,6,7,8-四氢-8-羟基喹啉(式(10)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应58min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为97%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,2-氯-5,6,7,8-四氢-8-羟基喹啉的分离收率为90%。
实施例21:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的6-甲氧基-1-己醇(式(11)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应120min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,6-甲氧基-1-己醇的分离收率为93%。
实施例22:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的1,6-己二醇单乙酸酯(式(12)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应90min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,1,6-己二醇单乙酸酯的分离收率为94%。
实施例23:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的1,6-己二醇单苯甲酸酯(式(13)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应100min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,1,6-己二醇单苯甲酸酯的分离收率为95%。
实施例24:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲氧基苄基保护的6-(甲氧基甲氧基)-1-己醇(式(14)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应100min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为98%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,6-(甲氧基甲氧基)-1-己醇的分离收率为93%。
实施例25:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol两个羟基均由4-甲氧基苄基保护的1,6-己二醇(式(15)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应120min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,1,6-己二醇的分离收率为96%。
实施例26:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol苄基保护的环己醇(式(16)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应200min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为98%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,环己醇的分离收率为92%。
实施例27:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 3,4-二甲氧基苄基保护的环己醇(式(17)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至140℃的油浴中,反应60min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,环己醇的分离收率为93%。
实施例28:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 3,4-二甲氧基苄基保护的环己醇(式(17)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至100℃的油浴中,反应240min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,环己醇的分离收率为93%。
实施例29:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 4-甲基苄基保护的环己醇(式(18)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应110min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为99%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,环己醇的分离收率为93%。
实施例30:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 3-甲基苄基保护的环己醇(式(19)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应150min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为100%,产物选择性为97%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,环己醇的分离收率为91%。
实施例31:
在300mL聚四氟乙烯内衬的压力釜中,加入8mmol 3-甲基苄基保护的环己醇(式(20)),10mL乙二醇二乙醚,0.4mmol的DDQ,0.4mmol的TBN,密闭压力釜,充氧气至压力表为0.2MPa,将压力釜放入到预先升温至120℃的油浴中,反应160min。降温并小心卸压后,有机相用气相色谱(GC)分析,转化率为93%,产物选择性为96%。过硅胶柱,以体积比为1∶3的乙酸乙酯和石油醚的混合物为洗脱剂,环己醇的分离收率为82%。
