
技术领域
本发明涉及有机半导体材料技术领域,尤其涉及一种工字形噻吩并苯化合物及其制备方 法。
背景技术
近些年,具有高载流子迁移率性质的有机半导体材料的发展异常活跃,在信息显示、集 成电路、光伏电池和传感器等方面显示出广阔的应用前景。有机场效应晶体管(OFETs)具 有有机半导体的诸多优良特性,如易于制备和功能化、较低的器件制备温度、良好的柔韧性、 和塑料衬底有良好的兼容性以及可大面积制备等,因而越来越受到人们的重视。
在OFETs器件中起关键作用的是有机半导体材料,按载流子传输特性可分为p型和n型 材料,其载流子分别为空穴和电子。其中,p型材料以并五苯为代表,发展较早。如今,因 为噻吩类化合物的良好的共轭性,较低的最高占据分子轨道(HOMO)以及较强的分子间作 用力(如硫-硫作用和碳-氢-π作用等)对于材料的空气稳定性和空穴传输较为有利,所以并 噻吩类材料已成为新的合成热点。
对于OFETs器件的蓬勃发展,目前却缺少足够的相关材料,无法满足日益增长的有机半 导体材料市场。
发明内容
本发明的目的是为了解决目前市场上并噻吩类材料的种类及量的缺乏,提供了一种工字 形噻吩并苯化合物。
为了达到上述发明目的,本发明采用以下技术方案:
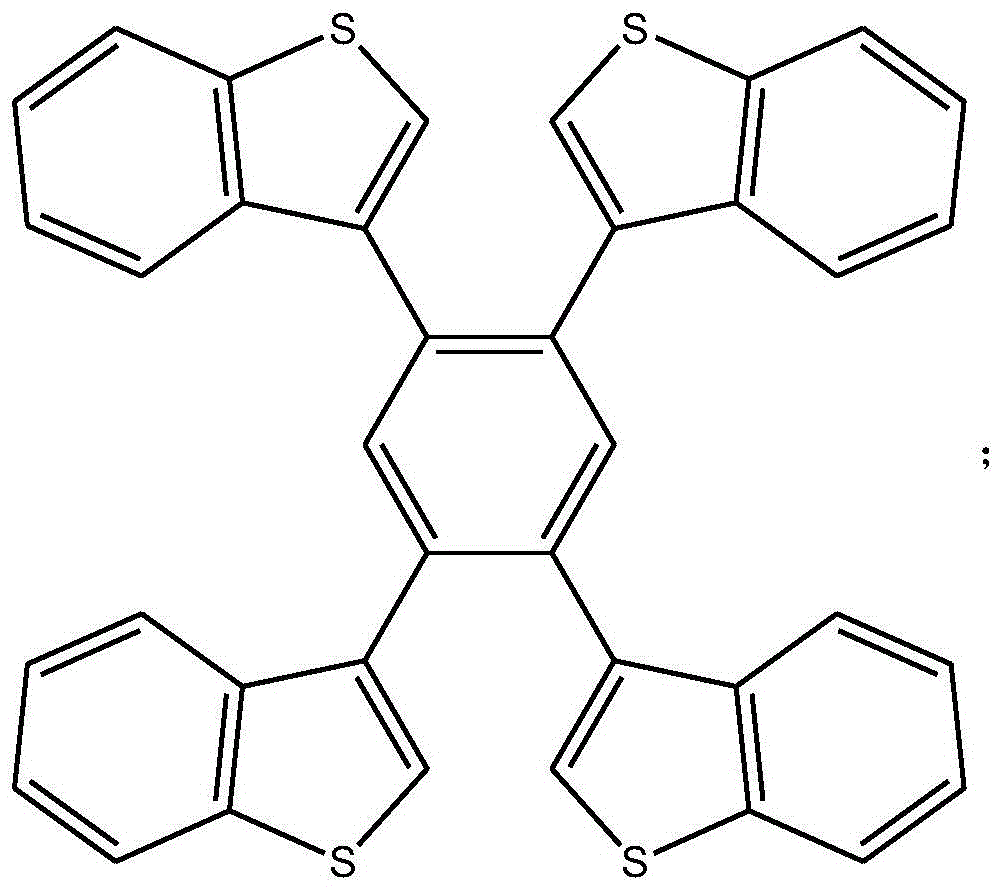
一种工字形噻吩并苯化合物,所述工字形噻吩并苯化合物的结构式为:
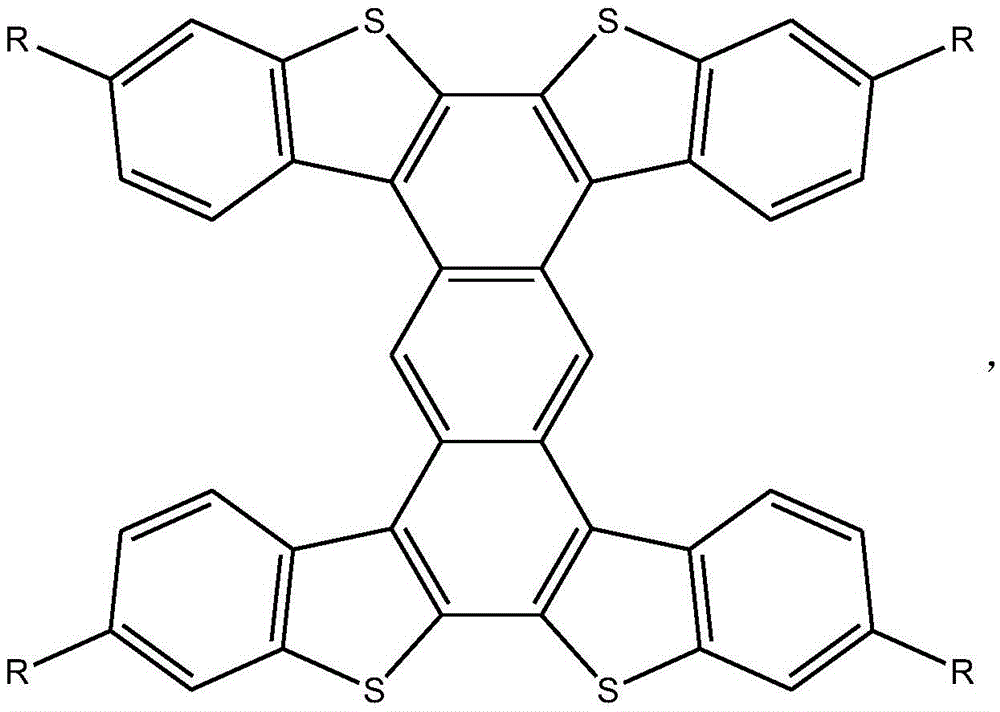
该结构式中的R为氢、烷基或芳基。
作为优选,所述工字形噻吩并苯化合物的结构式中的烷基的碳原子总数为1-20,芳基的 碳原子总数为6-30。
该烷基包括甲基、乙基、丙基、丙基、丁基、戊基、己基、庚基或者辛基等。芳基为苯 基、甲基苯基、乙基苯基等。
作为优选,所述结构式中的R为氢,所述工字形噻吩并苯化合物的结构式为:
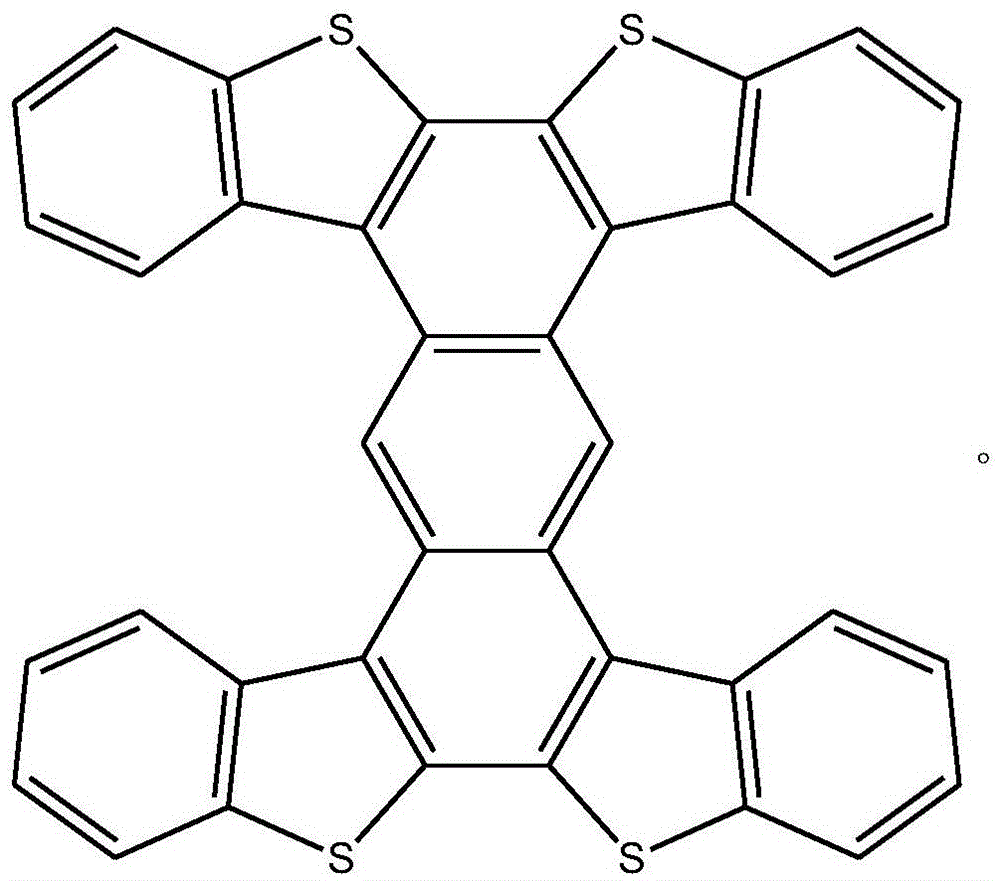
本发明的另一个目的是提供该化合物的制备方法,为了该发明目的,本发明采用以下技 术方案:
一种工字形噻吩并苯化合物的制备方法,该制备方法包括以下步骤:
(1)在氮气保护的条件下,将R取代的3-溴苯并噻吩先与有机锂试剂反应,生成苯并 噻吩锂盐,再加入异丙醇频哪醇硼酸酯,反应完毕得到R取代的苯并噻吩-3-硼酸频哪酯;反 应温度为-70--80℃,反应时间为20-24小时;
该步骤的温度优选为-78℃,反应时间优选为24h。
(2)在催化剂存在的条件下,将R取代的苯并噻吩-3-硼酸频哪酯与1,2,4,5-四溴苯 混匀进行Suzuki反应,反应完毕得到1,2,4,5-四(苯并噻吩基)苯化合物;所述1,2,4, 5-四溴苯、R取代的苯并噻吩-3-硼酸频哪酯和催化剂的摩尔比为1:4-8:0.1-0.2;所述催化剂为 四(三苯基膦)钯;反应温度为90-110℃,反应时间为15-30小时;
该步骤的反应温度优选为90℃,反应时间优选为24h。
(3)将步骤(2)所得1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂混匀进行氧化反 应,反应完毕得到工字形噻吩并苯化合物;1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂 的摩尔比为1:4-8;所述氧化剂为三氯化铁;所述氧化反应的反应温度为20-30℃,反应时间 为5-10小时。
本申请的制备方法中,
所述1,2,4,5-四溴苯、R取代的苯并噻吩-3-硼酸频哪酯和催化剂的摩尔比优选为 1:8:0.15。
所述1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂的摩尔比优选为为1:6;
本发明提供的工字形噻吩并苯化合物及其制备方法,首先,在无水无氧的低温条件下, R取代的3-溴苯并噻吩与硼酸脂反应,得到R取代的苯并噻吩-3-硼酸脂;然后,在催化剂存 在的条件下,将1,2,4,5-四溴苯与R取代的苯并噻吩-3-硼酸脂混匀进行Suzuki反应,反 应完毕得到1,2,4,5-四(苯并噻吩基)苯化合物;然后,将所得1,2,4,5-四(苯并噻 吩基)苯化合物与氧化剂混匀进行反应,反应完毕得到工字形噻吩并苯化合物;工字形噻吩 并苯化合物的合成路线简单、有效、原料为商业化产品,合成成本低、合成方法具有普适性, 可推广到其他各种取代基取代的工字形噻吩并苯化合物的合成,同时本发明合成的工字形噻 吩并苯化合物在场效应晶体管等有机半导体器件中有良好的应用前景,实用性强,具有较强 的推广与应用价值。
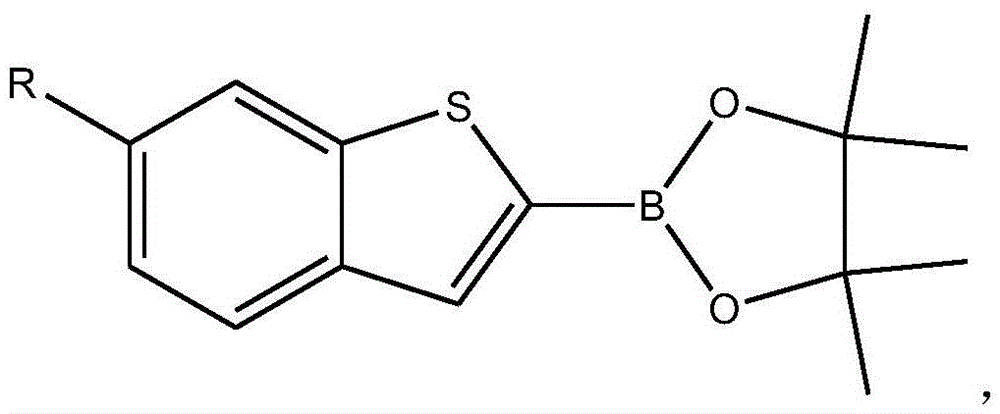
作为优选,所述R取代的苯并噻吩-3硼酸酯的结构式为:
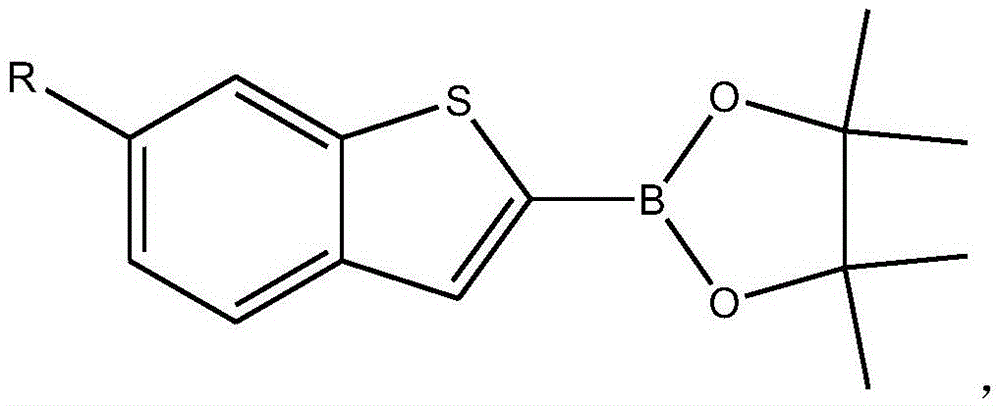
其中,所述R为氢、烷基或芳基。
作为优选,所述R为氢。
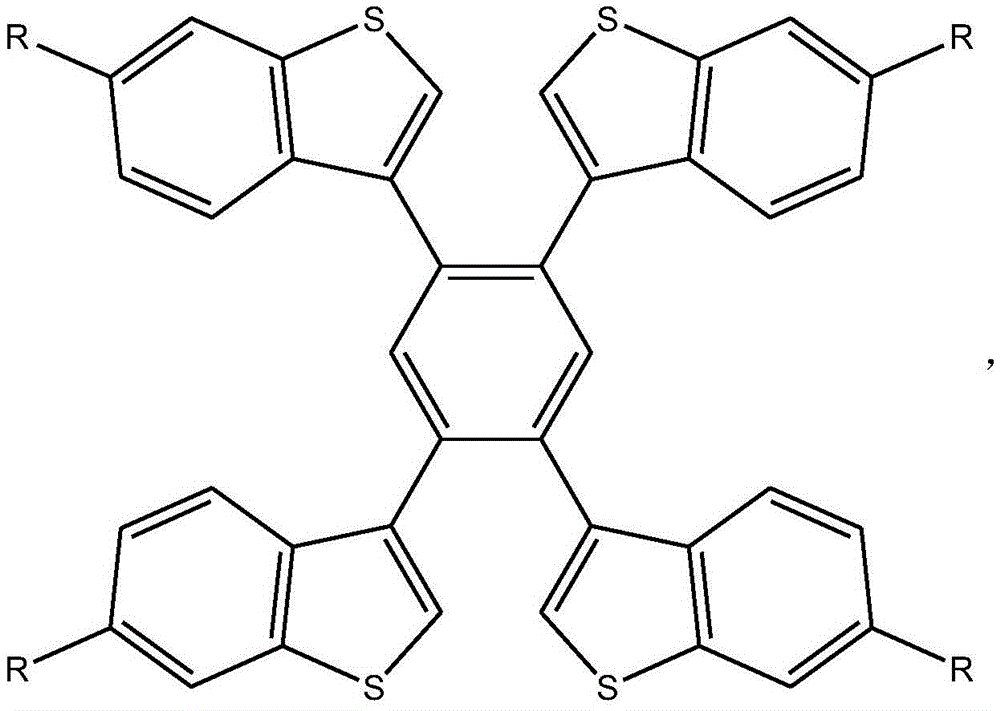
作为优选,在步骤(2)中,所述1,2,4,5-四(苯并噻吩基)苯化合物的结构式为:

其中,所述R为氢、烷基或芳基。
作为优选,在步骤(2)中,所述1,2,4,5-四(苯并噻吩基)苯化合物的取代基R为 氢,所述1,2,4,5-四(苯并噻吩基)苯化合物的结构式为:
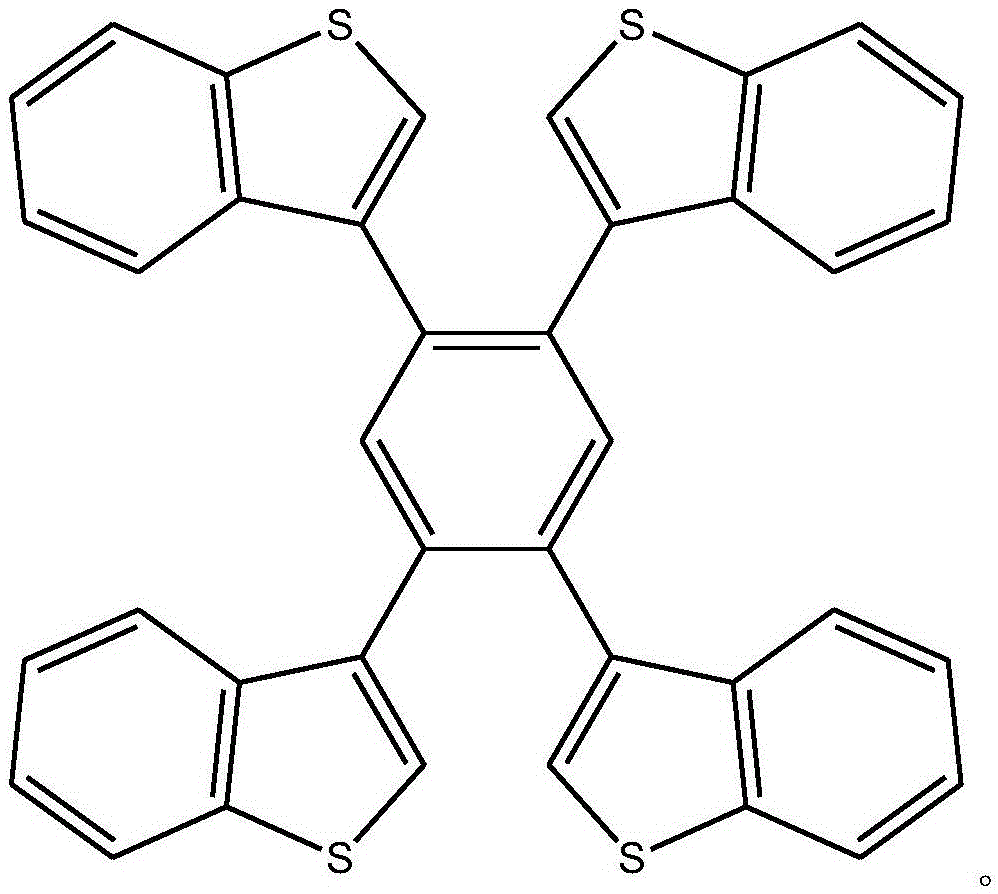
作为优选,在步骤(1)中,所述有机锂试剂为正丁基锂或叔丁基锂。
作为优选,步骤(2)和步骤(3)所进行的反应均在溶剂中进行,步骤(2)中的溶剂为 四氢呋喃,步骤(3)中的溶剂为二氯甲烷。
本发明与现有技术相比,有益效果是:
1、合成路线简单、有效;
2、原料为商业化产品,合成成本低、合成方法具有普适性,可推广到其他各种取代基取 代的工字形噻吩并苯化合物的合成;
3、本发明合成的工字形噻吩并苯化合物在场效应晶体管等有机半导体器件中有良好的半 导体性能,具有广泛的应用前景,实用性强,具有较强的推广与应用价值。
4、采用工字型结构,既可以在增加共轭结构单元以扩大电荷在分子内的离域范围的同时, 也可以避免稠环苯并噻吩类化合物之间的空间位阻,此外得到的平面分子间强的π-π相互作 用,有助于分子间形成紧密堆积,从而获得较高的电学性能。
附图说明
图1是本发明的制备方法中步骤(1)的合成路线图;
图2是本发明的制备方法中步骤(2)和步骤(3)的合成路线图;
图3是本发明制备的一种工字形噻吩并苯化合物的质谱图。
具体实施方式
下面通过具体实施例对本发明的技术方案作进一步描述说明。
如果无特殊说明,本发明的实施例中所采用的原料均为本领域常用的原料,实施例中所 采用的方法,均为本领域的常规方法。
实施例1:
一种工字形噻吩并苯化合物的制备方法包括以下步骤:(合成路线见图1和图2)
(1)在氮气保护的条件下,将3-溴苯并噻吩先与有机锂试剂-正丁基锂反应,生成苯并 噻吩锂盐,再加入异丙醇频哪醇硼酸酯,反应完毕得到苯并噻吩-3-硼酸频哪酯;反应温度为 -78℃,反应时间为24小时;
(2)在催化剂存在的条件下,将R取代的苯并噻吩-3-硼酸频哪酯与1,2,4,5-四溴苯 混匀进行Suzuki反应,反应完毕得到1,2,4,5-四(苯并噻吩基)苯化合物;所述1,2,4, 5-四溴苯、R取代的苯并噻吩-3-硼酸频哪酯和催化剂的摩尔比为1:4:0.15;所述催化剂为四(三 苯基膦)钯;反应温度为90℃,反应时间为24小时;反应在溶剂中进行,溶剂为四氢呋喃,
1,2,4,5-四(苯并噻吩基)苯化合物的结构式为:
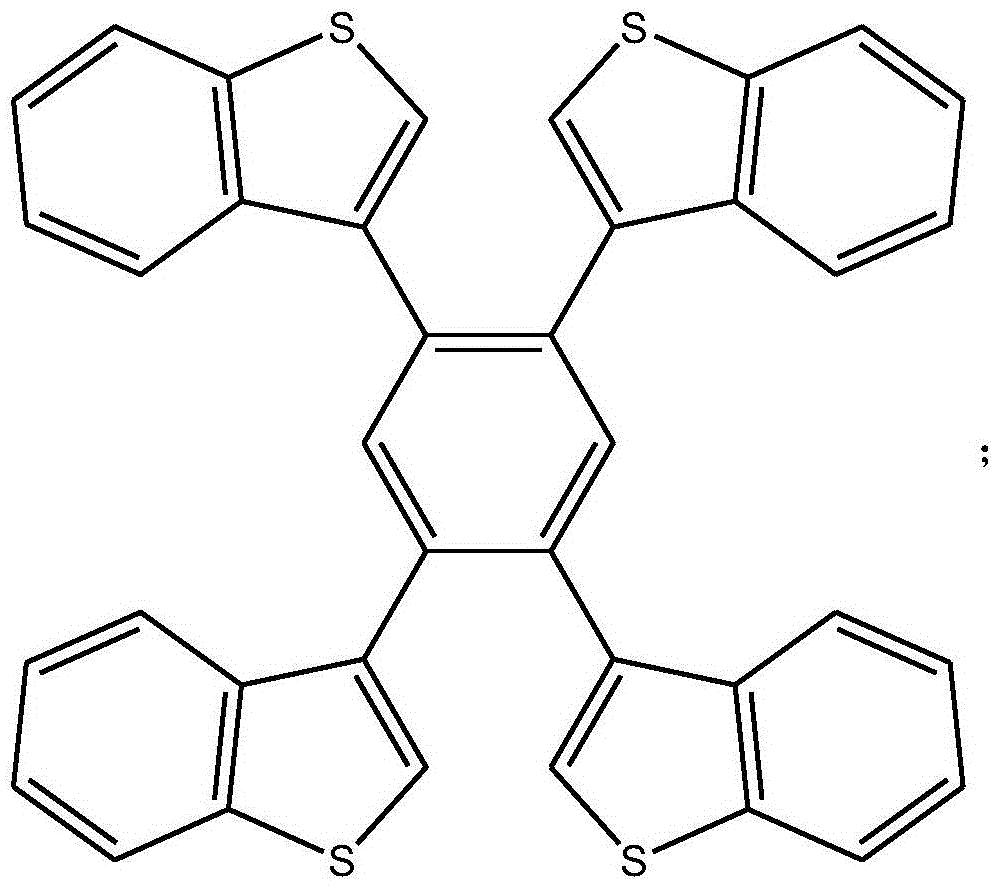
该步骤的具体操作为:在溶解有1,2,4,5-四溴苯的四氢呋喃溶液中(比例为1mmol 溶于100mL四氢呋喃),除气,加入Pd(PPh3)4,氩气中搅拌,加入苯并噻吩-3-硼酸,再加入 2Mol/L碳酸钠水溶液(1mmol的1,2,4,5-四溴苯加入10-20mL),升温至回流进行Suzuki 反应,反应24小时;冷却后直接过滤,用二氯甲烷萃、石油醚洗,经过氯苯重结晶得到白色 固体。
结构表征数据如下:
质谱:[MALID-TOF-MS]m/s606。
(3)将步骤(2)所得1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂混匀进行氧化反 应,反应完毕得到工字形噻吩并苯化合物;1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂 的摩尔比为1:4;所述氧化剂为三氯化铁;所述氧化反应的反应温度为20℃,反应时间为6 小时,反应在溶剂中进行,溶剂为二氯甲烷。
具体操作为:
将1,2,4,5-四(苯并噻吩基)苯化合物溶于干燥的二氯甲烷,搅拌,通入氮气,15 分钟后,慢慢将三氯化铁的硝基甲烷溶液滴加其中,反应6小时后,过滤,滤饼经水、乙醇、 四氢呋喃和二氯甲烷洗涤后,真空干燥。
得到的工字形噻吩并苯化合物的结构式为:
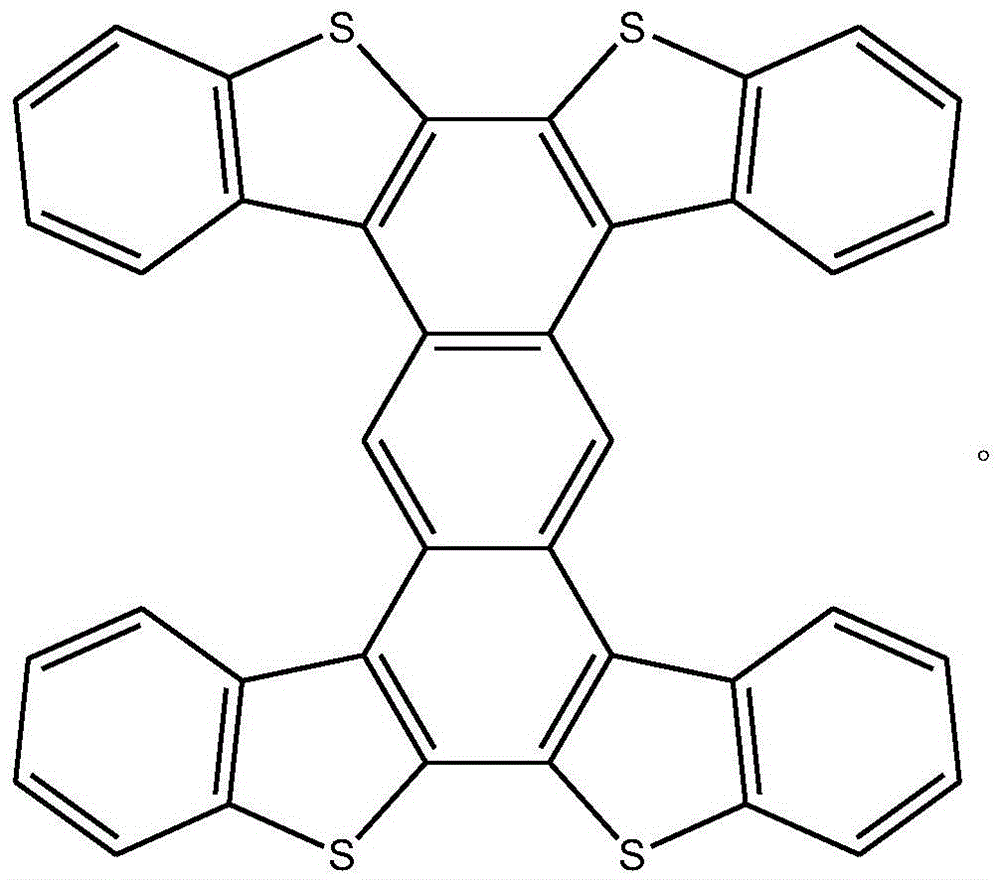
该工字形噻吩并苯化合物的质谱图见图3。
工字形噻吩并苯化合物的结构表征数据如下:
质谱:[HRMS]m/s602.0285;
元素分析:Anal.CalcdforC38H18S4:C,75.71;H,3.01;S,21.28.Found:C,75.48;H,3.00. 实施例2:
一种工字形噻吩并苯化合物的制备方法包括以下步骤:(合成路线见图1和图2)
(1)在氮气保护的条件下,将3-溴苯并噻吩先与有机锂试剂-叔丁基锂反应,生成苯并 噻吩锂盐,再加入异丙醇频哪醇硼酸酯,反应完毕得到苯并噻吩-3-硼酸频哪酯;反应温度为 -70℃,反应时间为20小时;
(2)在催化剂存在的条件下,将R取代的苯并噻吩-3-硼酸频哪酯与1,2,4,5-四溴苯 混匀进行Suzuki反应,反应完毕得到1,2,4,5-四(苯并噻吩基)苯化合物;所述1,2,4, 5-四溴苯、R取代的苯并噻吩-3-硼酸频哪酯和催化剂的摩尔比为1:8:0.1;所述催化剂为四(三 苯基膦)钯;反应温度为110℃,反应时间为15小时;反应在溶剂中进行,溶剂为四氢呋喃,
1,2,4,5-四(苯并噻吩基)苯化合物的结构式为:
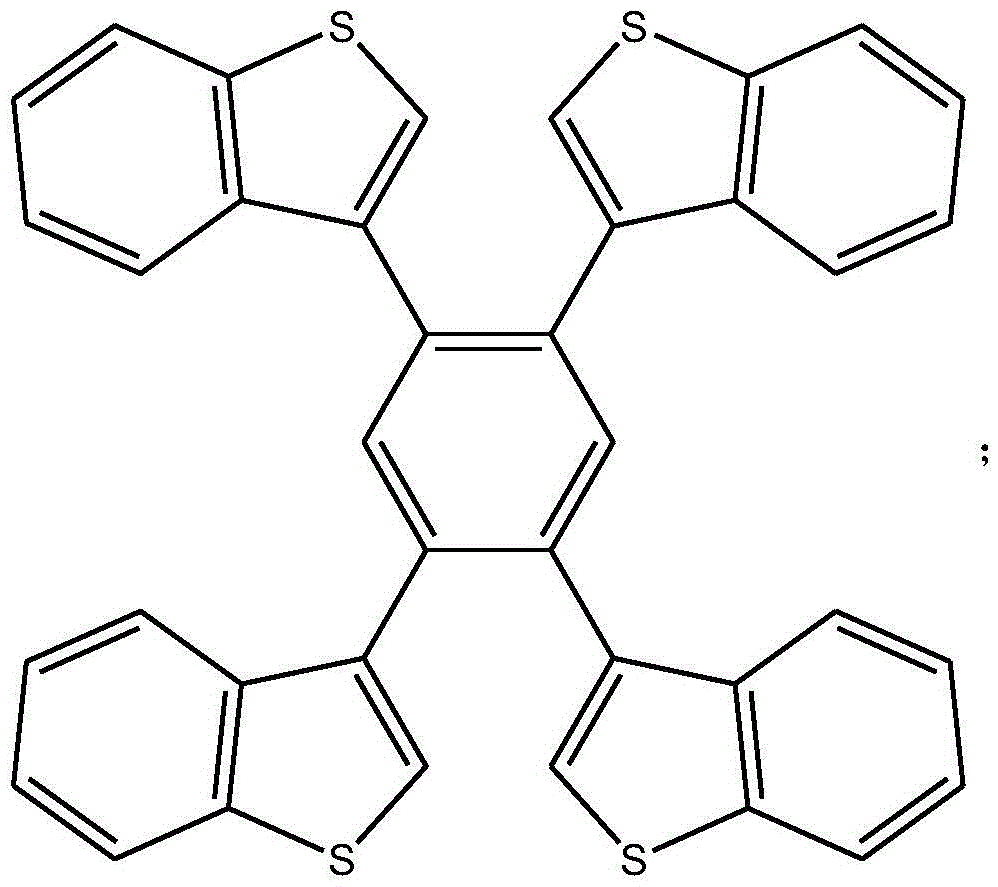
该步骤的具体操作为:在溶解有1,2,4,5-四溴苯的四氢呋喃溶液中(100mL四氢呋 喃溶解1mmol),除气,加入Pd(PPh3)4,氩气中搅拌,加入苯并噻吩-3-硼酸,再加入2Mol/L 碳酸钠水溶液(1mmol的1,2,4,5-四溴苯加入10-20mL),升温至回流进行Suzuki反应, 反应24小时;冷却后直接过滤,用二氯甲烷萃、石油醚洗,经过氯苯重结晶得到白色固体。
结构表征数据如下:
质谱:[MALID-TOF-MS]m/s606。
(3)将步骤(2)所得1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂混匀进行氧化反 应,反应完毕得到工字形噻吩并苯化合物;1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂 的摩尔比为1:8;所述氧化剂为三氯化铁;所述氧化反应的反应温度为30℃,反应时间为5 小时,反应在溶剂中进行,溶剂为二氯甲烷。
具体操作为:
将1,2,4,5-四(苯并噻吩基)苯化合物溶于干燥的二氯甲烷,搅拌,通入氮气,15 分钟后,慢慢将三氯化铁的硝基甲烷溶液滴加其中,反应5小时后,过滤,滤饼经水、乙醇、 四氢呋喃和二氯甲烷洗涤后,真空干燥。
得到的工字形噻吩并苯化合物的结构式为:
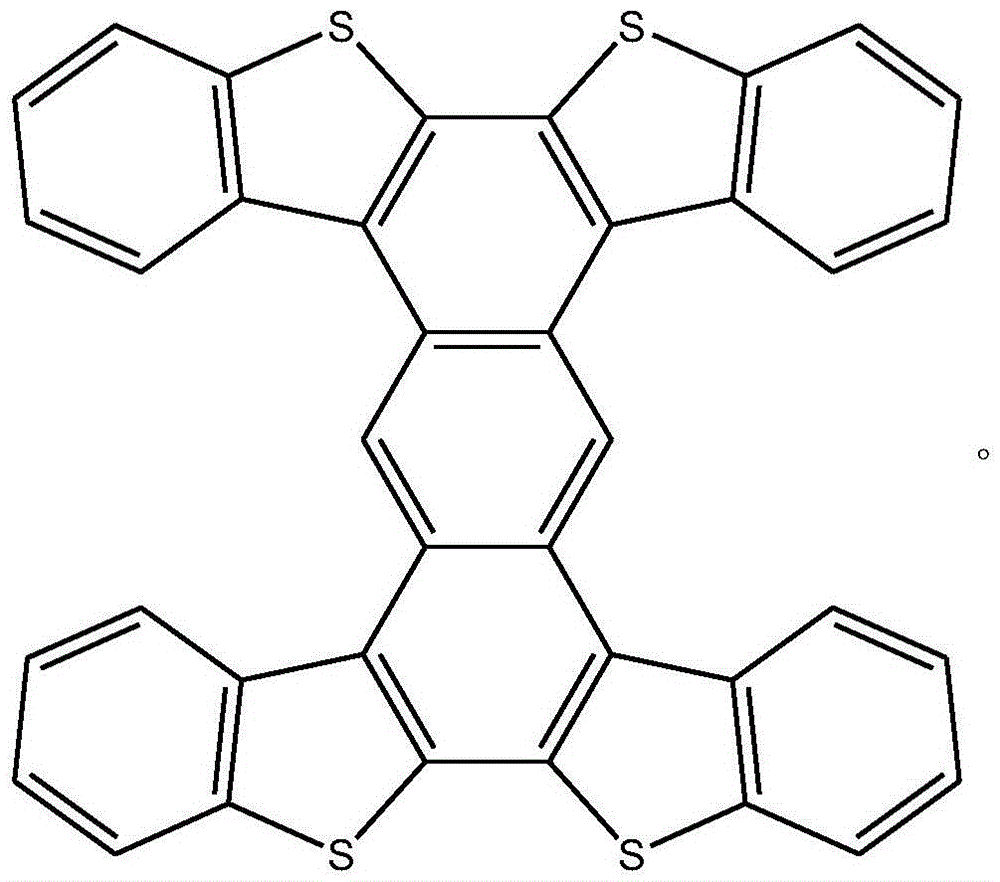
该工字形噻吩并苯化合物的质谱图见图3。
工字形噻吩并苯化合物的结构表征数据如下:
质谱:[HRMS]m/s602.0285;
元素分析:Anal.CalcdforC38H18S4:C,75.71;H,3.01;S,21.28.Found:C,75.48;H,3.00.
实施例3:
一种工字形噻吩并苯化合物的制备方法包括以下步骤:(合成路线见图1和图2)
(1)在氮气保护的条件下,将3-溴苯并噻吩先与有机锂试剂-正丁基锂反应,生成苯并 噻吩锂盐,再加入异丙醇频哪醇硼酸酯,反应完毕得到苯并噻吩-3-硼酸频哪酯;反应温度为 80℃,反应时间为20-24小时;
(2)在催化剂存在的条件下,将R取代的苯并噻吩-3-硼酸频哪酯与1,2,4,5-四溴苯 混匀进行Suzuki反应,反应完毕得到1,2,4,5-四(苯并噻吩基)苯化合物;所述1,2,4, 5-四溴苯、R取代的苯并噻吩-3-硼酸频哪酯和催化剂的摩尔比为1:4-8:0.1-0.2;所述催化剂为 四(三苯基膦)钯;反应温度为90-110℃,反应时间为15-30小时;反应在溶剂中进行,溶 剂为四氢呋喃,
1,2,4,5-四(苯并噻吩基)苯化合物的结构式为:
该步骤的具体操作为:在溶解有1,2,4,5-四溴苯的四氢呋喃溶液中(393mg,1mmol, 溶于100mL四氢呋喃),除气,加入150mgPd(PPh3)4,氩气中搅拌,加入苯并噻吩-3-硼酸 (1.4g,8.0mmol),再加入2Mol/L的碳酸钠水溶液20mL,升温至回流进行Suzuki反应,反 应24小时;冷却后直接过滤,用二氯甲烷萃、石油醚洗,经过氯苯重结晶得到白色固体400mg, 产率66%。
结构表征数据如下:
质谱:[MALID-TOF-MS]m/s606。
(3)将步骤(2)所得1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂混匀进行氧化反 应,反应完毕得到工字形噻吩并苯化合物;1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂 的摩尔比为1:4-8;所述氧化剂为三氯化铁;所述氧化反应的反应温度为20-30℃,反应时间 为5-10小时,反应在溶剂中进行,溶剂为二氯甲烷。
具体操作为:
将1,2,4,5-四(苯并噻吩基)苯化合物溶于干燥的二氯甲烷,搅拌,通入氮气,15 分钟后,慢慢将三氯化铁(1g)的硝基甲烷(5ml)溶液滴加其中,反应6小时后,过滤,滤 饼经水、乙醇、四氢呋喃和二氯甲烷洗涤后,真空干燥。
得到的工字形噻吩并苯化合物的结构式为:

该工字形噻吩并苯化合物的质谱图见图3。
工字形噻吩并苯化合物的结构表征数据如下:
质谱:[HRMS]m/s602.0285;
元素分析:Anal.CalcdforC38H18S4:C,75.71;H,3.01;S,21.28.Found:C,75.48;H,3.00. 实施例4:
一种工字形噻吩并苯化合物的制备方法,该制备方法包括以下步骤:
(1)在氮气保护的条件下,将R取代的3-溴苯并噻吩先与有机锂试剂反应,生成苯并 噻吩锂盐,再加入异丙醇频哪醇硼酸酯,反应完毕得到R取代的苯并噻吩-3-硼酸频哪酯;反 应温度为-70--80℃,反应时间为20-24小时;
所述R取代的苯并噻吩-3硼酸酯的结构式为:
(2)在催化剂存在的条件下,将R取代的苯并噻吩-3-硼酸频哪酯与1,2,4,5-四溴苯 混匀进行Suzuki反应,反应完毕得到1,2,4,5-四(苯并噻吩基)苯化合物;所述1,2,4, 5-四溴苯、R取代的苯并噻吩-3-硼酸频哪酯和催化剂的摩尔比为1:4-8:0.1-0.2;所述催化剂为 四(三苯基膦)钯;反应温度为90-110℃,反应时间为15-30小时;反应在溶剂中进行,溶 剂为四氢呋喃,
所述1,2,4,5-四(苯并噻吩基)苯化合物的结构式为:
(3)将步骤(2)所得1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂混匀进行氧化反 应,反应完毕得到工字形噻吩并苯化合物;1,2,4,5-四(苯并噻吩基)苯化合物与氧化剂 的摩尔比为1:4-8;所述氧化剂为三氯化铁;所述氧化反应的反应温度为20-30℃,反应时间 为5-10小时;反应在溶剂中进行,溶剂为二氯甲烷。
得到的工字形噻吩并苯化合物的结构式为:
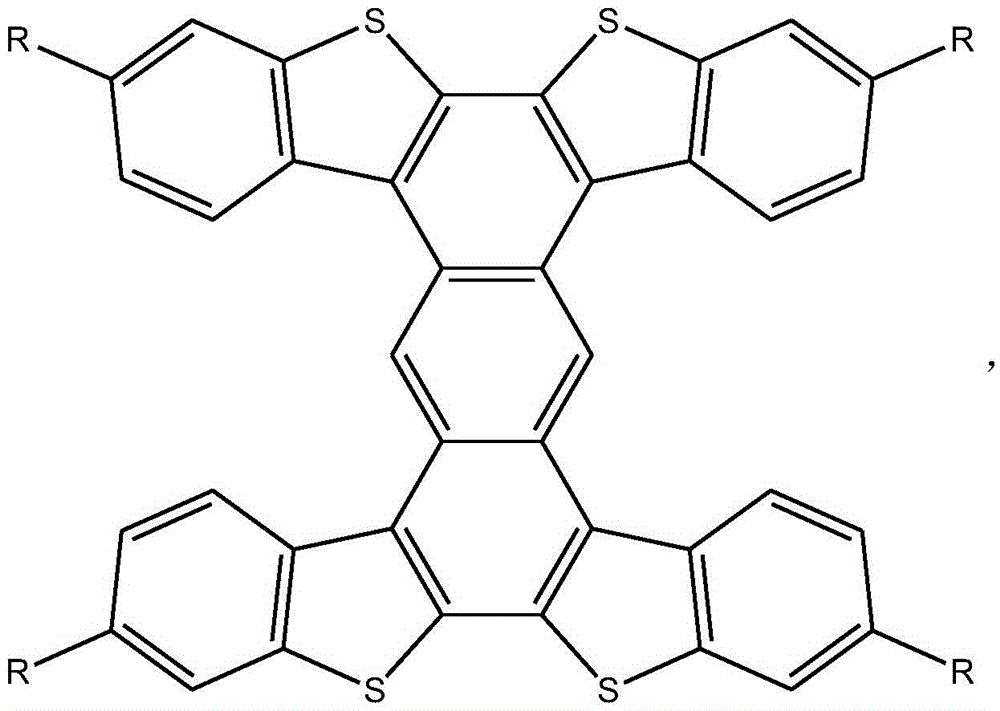
该结构式中的R为甲基。
实施例5:
制备方法与实施例4相同,R为辛基。
实施例6:
制备方法与实施例4相同,R为苯基。
实施例7:
制备方法与实施例4相同,R为甲苯基。
