
技术领域
本发明涉及有机化学领域,具体是一种含硅元素的化合物甲基苯基乙烯基乙氧基硅烷的合成方法。
背景技术
分子中含有1个可水解基团(如氯原子、烷氧基、酰氧基等)的有机硅化合物及其水解缩合生成的二聚体是有机硅材料表面重要的改性剂,也是控制有机硅聚合物分子量的重要封端剂。目前常用的有机硅类的封端剂主要包括六甲基二硅氧烷、四甲基二乙烯基二硅氧烷、二甲基苯基乙氧基硅烷等。
封端剂的组成及结构是影响有机硅聚合物性质如热性能、光学性能及机械力学性能的重要因素,例如,甲基二苯基硅氧基(MePh2SiO1/2)的耐热稳定性最好,甲基二苯基硅氧基封端的聚二甲基硅氧烷或聚(二甲基-甲基苯基)硅氧烷共聚物或聚(二甲基-二苯基)硅氧烷共聚物具有优异的热稳定性,可在大于350℃以上的高温环境中长期使用;因苯基的摩尔折射度远远大于甲基或乙烯基的摩尔折射度,因此使用含苯基的封端剂可以制备得到高折射率的MQ、MDQ、MTQ及MT等类型的有机硅树脂,对于提高白光LED封装材料的折射率及光学透明性具有重要的应用价值;封端剂分子结构中的硅乙烯基官能团属于活性官能团,它既可以在过氧化合物产生的自由基作用下发生自身交联或与其它含有乙烯基或炔键的化合物发生相互交联,也可以在铂、铑、钌、铁或铜等金属配合物催化剂作用下,与含有硅氢键的有机硅化合物或聚合物通过硅氢加成方式交联固化,形成网络状结构的有机硅聚合物材料,封端剂中的硅乙烯基官能团是影响有机硅聚合物材料机械性能的重要因素。
传统的有机硅聚合物封端剂主要分为全甲基类型的封端剂(如三甲基氯硅烷、三甲基烷氧基硅烷、六甲基二硅氧烷、八甲基三硅氧烷、十甲基四硅氧烷等)、甲基苯基类型封端剂(如甲基二苯基氯硅烷、甲基二苯基烷氧基硅烷、1,3-二甲基-1,1,3,3-四苯基二硅氧烷、二甲基苯基氯硅烷、二甲基苯基烷氧基硅烷、1,1,3,3-四甲基-1,3-二苯基二硅氧烷等)、羟基类型封端剂(如H2O、羟基硅油等)、活泼氢封端剂(如含氢双封头等)、活泼的乙烯基封端剂(如二甲基乙烯基氯硅烷、二甲基乙烯基烷氧基硅烷、1,1,3,3-四甲基-1,3-二乙烯基二硅氧烷等)、三氟丙基封端剂(如二甲基三氟丙基氯硅烷、二甲基三氟丙基烷氧基硅烷、1,1,3,3-四甲基-1,3-二(三氟丙基)二硅氧烷等)等。上述封端剂结构中的不同官能团赋予聚合物材料不同的物理化学性质,影响着材料的机械力学性能、光学性能、热性能、耐油、耐紫外光辐照及材料的加工性能。封端剂分子结构中同时含有多个功能性基团,则同时赋予有机硅聚合物材料具备相应的性能,如分子中同时含有苯基和乙烯基的甲基苯基乙烯基烷氧基硅烷或其二聚体(1,3-二甲基-1,3-二苯基-1,3-二乙烯基二硅氧烷),则既可以通过苯基赋予有机硅材料优异的光学性能和耐辐照性能,又可以通过乙烯基赋予有机硅材料优良的反应活性,进而通过乙烯基的交联固化或者与含硅氢键的化合物通过硅氢加成方式提高聚合物的机械力学性能。
中国发明专利CN104193775A公开了一种采用苯基格氏试剂制备含苯基乙烯基有机硅封端剂的方法,该方法在氮气保护和醚类溶剂中,利用氯苯或溴苯与金属镁反应先制备含苯基的格氏试剂,然后含苯基的格氏试剂在醚类溶剂中与含乙烯基的硅烷反应制备生成含苯基乙烯基的有机硅封端剂。该方法需要在无水无氧的条件下进行,制备格氏试剂时反应温度较高,且引发过程不易控制,增加了对设备和操作人员的要求,反应步骤多,降低了设备利用率。
发明内容
为了解决现有的格氏法制备甲基苯基乙烯基乙氧基硅烷存在反应条件苛刻,反应不易控制,副产物多,使用醚类溶剂,且醚类溶剂回收过程中因生成过氧化合物而容易发生爆炸危险等问题,本发明提出了一种甲基苯基乙烯基乙氧基硅烷的制备方法,反应路线短,反应过程温和,不需要苛刻的条件,以原料作为溶剂,工艺简单,非常适合大规模生产。
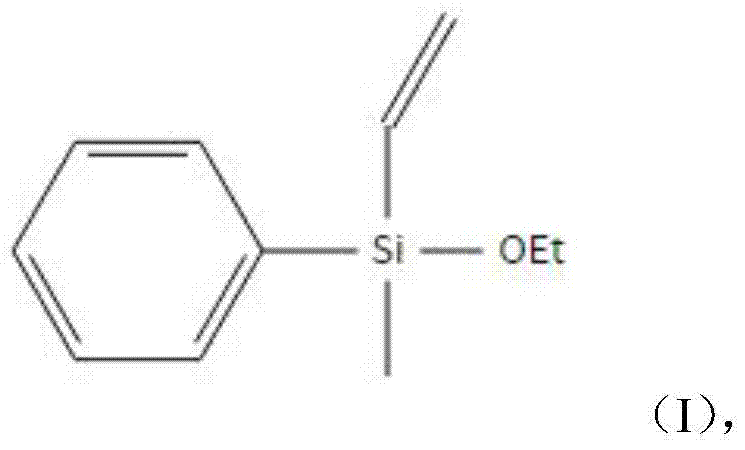
本发明是通过以下技术方案实现的:所述的甲基苯基乙烯基乙氧基硅烷以甲基乙烯基二乙氧基硅烷和氯苯为原料,通过钠缩合法制备,甲基苯基乙烯基乙氧基硅烷的结构式如(I)所示:
反应方程式如下式所示:

一种甲基苯基乙烯基乙氧基硅烷的制备方法为以下步骤:
(1)将甲基乙烯基二乙氧基硅烷和阻聚剂加入到容器中,搅拌下将温度升至为100~133℃,在搅拌的状态下加入钠块,并将钠块打成钠砂;作为优选,在115~133℃,将新切的钠片在机械搅拌作用下打成钠砂;
所述的阻聚剂选自烯类阻聚剂,作为优选,阻聚剂选自吩噻嗪、对苯二酚,使用量为甲基乙烯基二乙氧基硅烷质量的0.1%~15%,优选为0.1~6%。
(2)将氯苯滴加到反应体系中,控制反应温度115~133℃,氯苯滴加时间为0.5~10h,滴加完毕后维持反应0.1~20h;作为优选,氯苯的滴加时间为1~6h,滴加完毕维持反应0.5~6h;
甲基乙烯基二乙氧基硅烷与氯苯的摩尔比为1.0~6.0∶1,优选为2.0~4.0∶1;
金属钠与氯苯的摩尔比为1.5~4.0∶1。优选为2.0~3.8∶1;
(3)将步骤(2)得到产物冷却至0~50℃,优选冷却温度为15~40℃,滴加无水乙醇,然后再滴加甲基乙烯基二氯硅烷,甲基乙烯基二氯硅烷滴加时间控制在0.1~6h,优选滴加时间为1~4h,滴加完毕后维持反应1~8h,优选反应维持时间为1~3h,然后过滤除去固体杂质,收集滤液;作为优选,采用减压抽滤方式除去反应混合物中的固体杂质,同时收集滤液,滤液是包含本发明目标产物甲基苯基乙烯基乙氧基硅烷的混合溶液。
金属钠与无水乙醇的摩尔体积比为0.01~0.2mol∶1mL;
甲基乙烯基二氯硅烷的使用量为:每摩尔金属钠加入0.1~0.6mol氯原子。(4)对滤液进一步分离提纯得到甲基苯基乙烯基乙氧基硅烷。分离过程采用化工领域公知的精馏或减压精馏等分离提纯方法。
本发明以甲基乙烯基二乙氧基硅烷和氯苯为原料,通过钠缩合法制备甲基苯基乙烯基乙氧基硅烷,通过改变原料配比、反应温度、阻聚剂用量等参数,进一步提高目标产物收率。在钠缩合法合成甲基苯基乙烯基乙氧基硅烷中,甲基乙烯基二乙氧基硅烷既作为反应物又作为反应溶剂,将金属钠分散在甲基乙烯基二乙氧基硅烷中,形成钠砂,然后与氯苯发生缩合反应,得到含有甲基苯基乙烯基乙氧基硅烷的合成粗产物,合成的粗产物经公知的固液分离方法收集滤液,滤液再经公知的精馏等液液分离方法即可分离得到甲基苯基乙烯基乙氧基硅烷。
与现有技术相比,本发明的有益效果是:
(1)反应条件温和,副反应少,工艺简单;
(2)本发明克服了格氏法合成甲基苯基乙烯基乙氧基硅烷时需要苛刻的反应条件且使用易燃易爆的醚类溶剂等缺陷,降低了设备要求和操作的难度,非常适合于大规模工业生产。
具体实施方式
下面结合实施例对本发明作进一步详细说明,但实施例不是对本发明保护范围的限制,实施例中所用原料均为市购产品。
实施例1
在装有回流冷凝管、机械搅拌器及Pt-100铂电阻的干燥250mL四口烧瓶中,加入120.7g(0.753mol)甲基乙烯基二乙氧基硅烷和3.6g吩噻嗪,搅拌升温至124℃后加入新切的11.9g(0.517mol)钠片,将钠片打成钠砂;在3.5h内,将28.1g(0.250mol)氯苯通过恒压滴加漏斗滴加到反应体系中,控制反应温度在122~125℃之间,滴加完毕后维持反应2.5h,反应结束后冷却至25℃,慢慢加入3.0mL无水乙醇中和过量的钠;然后将18.99g甲基乙烯基二氯硅烷(含0.27mol的Cl原子)在2.5h内滴加到混合物中中和生成的醇钠;滴加完毕后,再在该温度下继续反应1h。然后将反应混合物减压抽滤,用少量甲基乙烯基二乙氧基硅烷淋洗滤饼,得到含有甲基苯基乙烯基乙氧基硅烷、甲基乙烯基二乙氧基硅烷及少量缩聚产物所组成的混合液体103.3g,经GC分析(面积归一法),混合液体中甲基苯基乙烯基乙氧基硅烷的含量为28.25%,目标产物收率为60.87%。
实施例2
在装有回流冷凝管、机械搅拌器及Pt-100铂电阻的干燥250mL四口烧瓶中,加入80.0g(0.499mol)甲基乙烯基二乙氧基硅烷和0.8g吩噻嗪,搅拌升温至115℃后加入新切的11.5g(0.5mol)钠片,将钠片打成钠砂;在6h内,将28.2g(0.250mol)氯苯通过恒压滴加漏斗滴加到反应体系中,控制反应温度在115~118℃之间,滴加完毕后维持反应1h,反应结束后冷却至15℃,慢慢加入3.2mL无水乙醇中和过量的钠;然后将19.2g甲基乙烯基二氯硅烷(含0.272mol的Cl原子)在4h内滴加到混合物中中和生成的醇钠;滴加完毕后,再在该温度下继续反应1h。然后将反应混合物减压抽滤,用少量甲基乙烯基二乙氧基硅烷淋洗滤饼,得到含有甲基苯基乙烯基乙氧基硅烷、甲基乙烯基二乙氧基硅烷及少量缩聚物所组成的混合液体88.3g,经GC分析(面积归一法),混合液体中甲基苯基乙烯基乙氧基硅烷的含量为24.75%,目标产物的收率为45.43%。
实施例3
在装有回流冷凝管、机械搅拌器及Pt-100铂电阻的干燥500mL四口烧瓶中,加入320.5g(1.999mol)甲基乙烯基二乙氧基硅烷和19.2g吩噻嗪,搅拌升温至133℃后加入新切的25.4g(1.104mol)钠片,将钠片打成钠砂;在1h内,将56.4g(0.501mol)氯苯通过恒压滴加漏斗滴加到反应体系中,控制反应温度在130~133℃之间,滴加完毕后维持反应0.5h,反应结束后冷却至30℃,慢慢加入6.5mL无水乙醇中和过量的钠;然后将38.19g甲基乙烯基二氯硅烷(含0.542mol的Cl原子)在1h内滴加到混合物中中和生成的醇钠;滴加完毕后,再在该温度下继续反应3h。然后将反应混合物减压抽滤,用少量甲基乙烯基二乙氧基硅烷淋洗滤饼,得到含有甲基苯基乙烯基乙氧基硅烷、甲基乙烯基二乙氧基硅烷及少量缩聚物所组成的混合液体220.5g,经GC分析(面积归一法),混合液体中甲基苯基乙烯基乙氧基硅烷的含量为30.70%,目标产物的收率为70.36%。
实施例4
在装有回流冷凝管、机械搅拌器及Pt-100铂电阻的干燥250mL四口烧瓶中,加入120.1g(0.749mol)甲基乙烯基二乙氧基硅烷和6.0g吩噻嗪,搅拌升温至115℃后加入新切的25.83g(1.124mol)钠片,将钠片打成钠砂;在2h内,将84.2g(0.749mol)氯苯通过恒压滴加漏斗滴加到反应体系中,控制反应温度在122~125℃之间,滴加完毕后维持反应6h,反应结束后冷却至40℃,慢慢加入112.4mL无水乙醇中和过量的钠;然后将19.2g甲基乙烯基二氯硅烷(含0.272mol的Cl原子)在1h内滴加到混合物中中和生成的醇钠;滴加完毕后,再在该温度下继续反应1h。然后将反应混合物减压抽滤,用少量甲基乙烯基二乙氧基硅烷淋洗滤饼,得到含有甲基苯基乙烯基乙氧基硅烷、甲基乙烯基二乙氧基硅烷及少量缩聚物所组成的混合液体95.6g,经GC分析(面积归一法),混合液体中甲基苯基乙烯基乙氧基硅烷的含量为25.15%,目标产物的收率为50.21%。
实施例5
在装有回流冷凝管、机械搅拌器及Pt-100铂电阻的干燥500mL四口烧瓶中,加入320.6g(2.0mol)甲基乙烯基二乙氧基硅烷和16.1g吩噻嗪,搅拌升温至120℃后加入新切的28.53g(1.24mol)钠片,将钠片打成钠砂;在1h内,将37.6g(0.334mol)氯苯通过恒压滴加漏斗滴加到反应体系中,控制反应温度在130~133℃之间,滴加完毕后维持反应3h,反应结束后冷却至25℃,慢慢加入6.2mL无水乙醇中和过量的钠;然后将9.6g甲基乙烯基二氯硅烷(含0.136mol的Cl原子)在1h内滴加到混合物中中和生成的醇钠;滴加完毕后,再在该温度下继续反应3h。然后将反应混合物减压抽滤,用少量甲基乙烯基二乙氧基硅烷淋洗滤饼,得到含有甲基苯基乙烯基乙氧基硅烷、甲基乙烯基二乙氧基硅烷及少量缩聚物所组成的混合液体230.6g,经GC分析(面积归一法),混合液体中甲基苯基乙烯基乙氧基硅烷的含量为27.80%,目标产物的收率为67.23%。
将实施例1~实施例5经过固液分离得到的液体混合物混合,然后经常压蒸馏回收甲基乙烯基二乙氧基硅烷后,再进行减压精馏,得到纯度为98.5%的甲基苯基乙烯基乙氧基硅烷160.8g,对产物进行GC-MS(TRACEDSQ)及1H-NMR(Burker,400MHz)分析,以确定和验证目标化合物的结构。
波谱分析结果如下:
GC-MSm/z(%):192(M+),177(100),163(36),137(20),133(49),73(21)。
1H-NMR(CDCl3):-Si-CH3(0.2ppm),-SiOCH2CH3(-CH3,1.2ppm),-Si-OCH2CH3(-CH2-,3.6ppm),-Si-Vi(5.5~6.2ppm),-Si-C6H5(7.0~7.6ppm)。
