
背景技术
本发明涉及有机高分子化合物技术领域,具体地说涉及一种光学活性聚硅烷的制备方法。
背景技术
光学活性聚硅烷是一类由Si-Si单键构筑的螺旋主链及连接在主链上的手性或非手性取代基团所组成的具有稳定的单向螺旋结构的聚合物。由于分子内存在σ-共轭效应,使电子可以沿着主链广泛离域,因此这类聚硅烷有着其它碳系、硅氧系聚合物所没有的特殊物理、化学性质,是制备手性传感器、分析器、放大器、手性光学开关及手性光学存储器的理想材料。近来研究发现光学活性聚硅烷在分子识别和作为手性固定相上也有着广泛的应用。目前,现有技术中一般采用由端基引入手性基团诱导聚硅烷的螺向选择,因此生成光学活性聚硅烷的制备方法大致有两种:a)以(R)或(S)-2-苯丙基氯硅烷进行手性封端[J. Am. Chem. Soc., 119(1997), 11345];b)以薄荷醇钾进行手性封端[Bull. Chem. Soc. Jpn., 77, (2004),1607];但是,方法a)的手性封端硅烷由手性的卤代烷烃合成得到,具有成本高,制备困难的特点;方法b) 使用掩蔽二硅烯方法制备聚硅烷,这种方法聚合物前体制备繁琐困难,而且薄荷醇钾的制备需要使用金属钾,由于金属钾反应活性高,容易燃烧和爆炸,进行大规模的生产面临着很大的困难。
发明内容
为了解决现有技术中生成光学活性聚硅烷的两种制备方法中存在的问题,本发明提供了一种光学活性聚硅烷的制备方法,本发明提供一种操作方便,在温和的条件下,以手性醇淬灭反应的方法制备光学活性聚硅烷。
本发明是通过以下技术方案实现的:一种光学活性聚硅烷的制备方法,在有机溶剂中以二氯硅烷为原料, Sm和SmI2为还原剂,手性醇R3OH做封端剂合成光学活性聚硅烷, 其反应式为:
 ;
;
式中:R1,R2分别选自烷基或者芳基;R3 与手性醇R3OH的R3一致,手性醇R3OH选自下式中的一种:
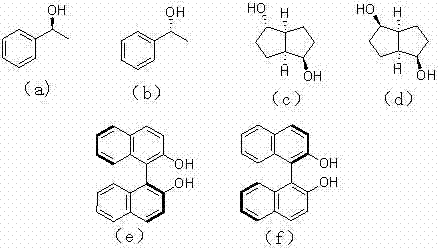 ;
;
光学活性聚硅烷中n为10-1500的整数;
有机溶剂选自四氢呋喃、1,2-二甲氧基乙烷、苯、甲苯中的一种。
作为优选,反应式中R1,R2分别选自 CH3、(CH2)xCH3 、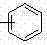 、
、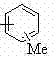 中一种,其中X = 1~5。
中一种,其中X = 1~5。
一种光学活性聚硅烷的制备方法,所述制备方法的具体步骤为:
(1)分别称取二氯硅烷、钐粉Sm、1,2-二碘乙烷、手性醇和有机溶剂,二氯硅烷、钐粉Sm、1,2-二碘乙烷、手性醇的摩尔质量比为1:4~8:1~2:0.05~1,有机溶剂为1mmol二氯硅烷称取10~250mL有机溶剂;有机溶剂为纯溶剂。
(2)在容器中加入步骤(1)中称取的钐粉Sm、1,2-二碘乙烷和有机溶剂,在惰性气体保护下反应1~2小时,制备出混合还原剂钐粉Sm和SmI2;作为优选,先抽真空再充惰性气体,惰性气体优选自氮气或氩气。
(3)然后向步骤(2)体系中加入步骤(1)中称取的二氯硅烷,并加热回流1~24小时,冷却至室温后,再加入步骤(1)中称取的手性醇,充分搅拌,得到光学活性聚硅烷粗品;
(4)对步骤(3)的光学活性聚硅烷粗品用萃取剂进行萃取,有机层经洗涤后干燥、过滤、蒸去萃取剂,最后再用溶解剂溶解后,加液体醇再沉淀,即得到光学活性聚硅烷;所述的萃取剂选自乙醚、甲苯、乙酸乙酯、氯仿或二氯甲烷,萃取剂的量为1毫摩尔二氯硅烷萃取剂为10-50毫升;溶解剂选自四氢呋喃、1,2-二甲氧基乙烷、苯、乙醚、氯仿、二氯甲烷或甲苯,溶解剂的量为使溶质完全溶解的量,液体醇选自乙醇或甲醇,液体醇的量为沉淀不再产生的量。
本发明在现有的各种合成光学活性聚硅烷的基础上,经过反复试验研究,发现在以二氯硅烷为原料, Sm/SmI2为还原聚集剂制备聚硅烷时,手性醇可以和聚硅烷端基的硅氯键进行反应,实现对聚硅烷的手性封端,进而诱导聚硅烷的螺向选择,生成光学活性的聚硅烷。
圆二色性(CD)光谱图,紫外光谱(UV)图证实了这一点。如图1所示,在聚硅烷最大紫外吸收处(340 nm),主链形成的吸收峰,无规聚硅烷没有CD吸收,而诱导后的光学活性聚硅烷呈现正的卡藤效应(Cotton Effect)。
与现有技术相比,本发明的有益效果是:
a) 步骤短,操作简便,反应条件比较温和,避免了工业危险品金属钾的使用。
b) 原料为商品化试剂,廉价易得,无需制备手性氯硅烷试剂。
附图说明
图1:圆二色性(CD)光谱图与紫外光谱(UV)图所示的卡藤效应;
其中,1:光学活性聚硅烷固体圆二色性光谱图;2:无规聚硅烷固体圆二色性光谱图;3:光学活性聚硅烷紫外光谱图;4:光学活性聚硅烷荧光光谱图。
具体实施方式
下面通过实施例,对本发明的技术方案作进一步具体的说明,但实施例不是对本发明保护范围的限制,还可以有许多的操作组合,本领域的普通技术人员能够从下述实例中直接导出到所有的情形,均应当是本发明的保护范围。
实施例1
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(1.2 g, 8 mmol) 金属钐粉,(0.56 g, 2 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入20 mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(0.38 g,2 mmol)苯基甲基二氯硅烷,加热回流24小时后,冷却至室温,加入(0.37 g,3 mmol)S-1-苯基乙醇,结构式如手性醇(a)所示,搅拌1小时。反应结束后,用乙醚萃取(3×20 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(2 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚甲基苯基硅烷。
聚甲基苯基硅烷平均分子量分布为Mw/Mn = 1.92,平均分子量为Mn =4682,收率为26%。 紫外光谱的最大吸收峰为340 nm。
实施例2
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(0.6g, 4 mmol) 金属钐粉,(0.28 g, 1mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入10 mL新蒸馏的1,2-二甲氧基乙烷溶剂,在氮气保护下,室温搅拌2小时,形成蓝黑色的SmI2。加入(0.21 g,1 mmol)苯基乙基二氯硅烷,加热回流12小时后,冷却至室温,加入(0.15 g,1.5 mmol)S-1-苯基乙醇,结构式如手性醇(a)所示,搅拌1小时。反应结束后,用乙醚萃取(3×10 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×5 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水甲苯(2 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚乙基苯基硅烷。
聚乙基苯基硅烷平均分子量分布为Mw/Mn = 1.92,平均分子量为Mn =3446,收率为25%。紫外光谱的最大吸收峰为342 nm。
实施例3
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的250 mL三口圆底烧瓶中放置(2.4 g, 16 mmol) 金属钐粉,(1.12 g, 4 mmol) 1,2-二碘乙烷抽真空,充氩气反复进行3次。然后注入100 mL新蒸馏的甲苯溶剂,在氩气保护下,室温搅拌2小时,形成蓝黑色的SmI2。加入(0.76 g,4 mmol)苯基丙基二氯硅烷,加热回流20小时后,冷却至室温,加入(0.47 g,4 mmol)S-1-苯基乙醇结构式如手性醇(a)所示,搅拌1小时。反应结束后,用乙醚萃取(3×50 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×20 mL), 饱和食盐水溶液洗涤三次(3×20 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(5 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚苯基丙基硅烷。
聚苯基丙基硅烷平均分子量分布为Mw/Mn = 1.73,平均分子量为Mn =6966,收率为33%。紫外光谱的最大吸收峰为345 nm。
实施例4
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(1.8 g, 12 mmol) 金属钐粉,(0.84 g, 3 mmol) 1,2-二碘乙烷抽真空,充氩气反复进行3次。然后注入30mL(蒸馏的四氢呋喃溶剂,在氩气保护下,室温搅拌1.5小时,形成蓝黑色的SmI2。加入(3 mmol)甲基正己基二氯硅烷,加热回流24小时后,冷却至室温,加入(0.16 g,1 mmol) Isomannide ,结构式如手性醇(d)所示,搅拌1小时。反应结束后,用乙酸乙酯萃取(3×30 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用二氯甲烷((2 mL)溶解后,用无水甲醇再沉淀,得到光学活性的聚甲基正己基硅烷。
聚甲基正己基硅烷平均分子量分布为Mw/Mn = 1.92,平均分子量为Mn = 4682,收率为28%。紫外光谱的最大吸收峰为330 nm。
实施例5
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50mL三口圆底烧瓶中放置1.8g,12mmol金属钐粉,(0.56 g, 2 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入50 mL新蒸馏的1,2-二甲氧基乙烷,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(0.38 g,2 mmol)苯基对甲苯基二氯硅烷,加热回流24小时后,冷却至室温,加入(0.57 g,2 mmol)S-联萘酚,结构式如手性醇(e)所示,搅拌1小时。反应结束后,用乙醚萃取(3×20 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无乙醚(2 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚苯基对甲苯基硅烷。
聚苯基对甲苯基硅烷平均分子量分布为Mw/Mn = 2.32,平均分子量为Mn = 5685,收率为45%。紫外光谱的最大吸收峰为351 nm,。
实施例6
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的1000 mL三口圆底烧瓶中放置(1.8g,12mmol) 金属钐粉,(2.8g,10 mmol)1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入200 mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌2小时,形成蓝黑色的SmI2。加入(2mmol)二己基二氯硅烷,加热回流24小时后,冷却至室温,加入(8.6 g,1mmol) R-联萘酚,结构式如手性醇(f)所示,搅拌1小时。反应结束后,用乙醚萃取(3×200 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×100 mL), 饱和食盐水溶液洗涤三次(3×100 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水苯(20 mL)溶解后,用无水甲醇再沉淀,得到光学活性的聚二己基硅烷。
聚二己基硅烷平均分子量分布为Mw/Mn = 2.52,平均分子量为Mn = 3258,收率为31%。紫外光谱的最大吸收峰为332 nm。
实施例7
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50mL三口圆底烧瓶中放置3.6g,24mmol 金属钐粉,(1.68g,6mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入50mL新蒸馏的四氢呋喃溶剂(甲苯),在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入3mmol苯基正己基二氯硅烷,加热回流24小时后,冷却至室温,加入(0.37 g,3 mmol)R-1-苯基乙醇,结构式如手性醇(b)所示,搅拌1小时。反应结束后,用氯仿萃取(3×20 mL),有机层用饱和碳酸氢钠洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水甲苯(2 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚正己基苯基硅烷。
聚正己基苯基硅烷平均分子量分布为Mw/Mn = 182,平均分子量为Mn = 6254,收率为28%。紫外光谱的最大吸收峰为344 nm。
实施例8
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的250mL三口圆底烧瓶中放置(1.8 g, 12 mmol) 金属钐粉,(0.42 g, 3 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入50mL新蒸馏的苯溶剂,在氮气保护下,室温搅拌1.5小时,形成蓝黑色的SmI2。加入(3 mmol)乙基邻甲苯基二氯硅烷,加热回流1小时后,冷却至室温,加入(0.18 g,2 mmol)R-1-苯基乙醇,结构式如手性醇(b)所示,搅拌1小时。反应结束后,用二氯甲烷萃取(3×30 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水1,2-二甲氧基乙烷(3 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚乙基邻甲苯基硅烷。
聚乙基邻甲苯基硅烷平均分子量分布为Mw/Mn = 2.32,平均分子量为Mn = 3846,收率为25%。紫外光谱的最大吸收峰为341 nm。
实施例9
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(1.5g,10mmol)金属钐粉,(0.84,3mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入20 mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(2 mmol)苯基正戊基二氯硅烷,加热回流18小时后,冷却至室温,加入(0.3mmol)D-isosorbide,结构式如手性醇(c)所示,搅拌1小时。反应结束后,用甲苯萃取(3×20mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用氯仿(3 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚苯基正戊基硅烷。
聚苯基正戊基硅烷平均分子量分布为Mw/Mn = 1.72,平均分子量为Mn = 5426,收率为32%。紫外光谱的最大吸收峰为343 nm。
实施例10
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的25 mL三口圆底烧瓶中放置(1.2g,8mmol) 金属钐粉,(0.48g,1.5mmol)1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入30 mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(1mmol)苯基间甲苯基二氯硅烷,加热回流8小时后,冷却至室温,加入(0.11 g,0.75 mmol)Isomannide,结构式如手性醇(d)所示,搅拌1小时。反应结束后,用乙醚萃取(3×10 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×5 mL), 饱和食盐水溶液洗涤三次(3×5 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水甲苯(0.5 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚苯基间甲苯基硅烷。
聚苯基间甲苯基硅烷平均分子量分布为Mw/Mn = 1.82,平均分子量为Mn = 5536,收率为31%。紫外光谱的最大吸收峰为351 nm。
实施例11
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(0.6 g, 4 mmol) 金属钐粉,(0.28 g, 1 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入20 mL新蒸馏的苯溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(1 mmol)乙基正丁基二氯硅烷,加热回流24小时后,冷却至室温,加入(1 mmol)S-联萘酚,结构式如手性醇(e)所示,搅拌1小时。反应结束后,用乙醚萃取(3×20 mL),有机层用饱和碳酸氢钠洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(1 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚乙基正丁基硅烷。
聚乙基正丁基硅烷平均分子量分布为Mw/Mn = 2.52,平均分子量为Mn = 3546,收率为25%。紫外光谱的最大吸收峰为328 nm。
实施例12
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的250 mL三口圆底烧瓶中放置(6.4g,35mmol)金属钐粉,(2.8 g, 10 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入80 mL新蒸馏的四氢呋喃溶剂,在氩气保护下,室温搅拌2小时,形成蓝黑色的SmI2。加入(5 mmol)对甲苯基乙基二氯硅烷,加热回流24小时后,冷却至室温,加入(4 mmol)R-联萘酚,结构式如手性醇(f)所示,搅拌1小时。反应结束后,用乙醚萃取(3×100mL),有机层用饱和碳酸氢钠洗涤二次(2×110 mL), 饱和食盐水溶液洗涤三次(3×100 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(10 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚对甲苯基乙基硅烷。
聚对甲苯基乙基硅烷平均分子量分布为Mw/Mn = 1.72,平均分子量为Mn = 5635,收率为35%。紫外光谱的最大吸收峰为342 nm。
实施例13
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(1.2 g, 8 mmol) 金属钐粉,(0.56 g, 2 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入20 mL新蒸馏的甲苯溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(2 mmol)苯基甲基二氯硅烷,加热回流24小时后,冷却至室温,加入(1mmol) D-isosorbide,结构式如手性醇(c)所示,搅拌1小时。反应结束后,用乙醚萃取(3×20 mL),有机层用饱和碳酸氢钠洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水苯(2 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚甲基苯基硅烷。
聚甲基苯基硅烷平均分子量分布为Mw/Mn = 2.21,平均分子量为Mn = 5682,收率为36%。紫外光谱的最大吸收峰为338 nm。
实施例14
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50mL三口圆底烧瓶中放置(1.8 g, 12 mmol) 金属钐粉,(0.84 g, 3 mmol) 1,2-二碘乙烷,抽真空充氩气反复进行3次。然后注入25 mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(2 mmol)正二丁基二氯硅烷,加热回流18小时后,冷却至室温,加入(2 mmol)R-1-苯基乙醇,结构式如手性醇(b)所示,搅拌1小时。反应结束后,用乙醚萃取(3×20mL),有机层用饱和碳酸氢钠洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(2 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚正二丁基硅烷。
聚正二丁基硅烷平均分子量分布为Mw/Mn = 2.73,平均分子量为Mn = 3966,收率为24%。紫外光谱的最大吸收峰为328 nm。
实施例15
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的250 mL三口圆底烧瓶中放置(2.4 g, 16 mmol) 金属钐粉,(2.16g, 4 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入50 mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌2小时,形成蓝黑色的SmI2。加入(3 mmol)苯基正丁基二氯硅烷,加热回流24小时后,冷却至室温,加入(0.3 mmol)D-isosorbide,结构式如手性醇(c)所示,搅拌1小时。反应结束后,用乙醚萃取(3×30 mL),有机层用饱和碳酸氢钠洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(3 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚苯基正丁基硅烷。
聚苯基正丁基硅烷平均分子量分布为Mw/Mn = 1.83,平均分子量为Mn = 5766,收率为31%。紫外光谱的最大吸收峰为342 nm。
实施例16
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(1.2 g, 8 mmol) 金属钐粉,(0.56 g, 2 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入100mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(0.38 g,2 mmol)丙基丁基二氯硅烷,加热回流24小时后,冷却至室温,加入(1.5 mmol)Isomannide ,结构式如手性醇(d)所示,搅拌1小时。反应结束后,用甲苯萃取(3×20 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(2 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚丙基丁基硅烷。
聚丙基丁基硅烷平均分子量分布为Mw/Mn = 2.53,平均分子量为Mn = 3546,收率为22%。紫外光谱的最大吸收峰为328 nm。
实施例17
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(0.75g,5mmol)金属钐粉,(0.28 g, 1 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入20 mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(1 mmol)对甲苯基丙基二氯硅烷,加热回流24小时后,冷却至室温,加入(0.05 mmol)S-联萘酚,结构式如手性醇(e)所示,搅拌1小时。反应结束后,用乙醚萃取(3×10 mL),有机层用饱和碳酸氢钠洗涤二次(2×5 mL), 饱和食盐水溶液洗涤三次(3×5 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(1 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚对甲苯基丙基硅烷。
聚对甲苯基丙基硅烷平均分子量分布为Mw/Mn = 1.33,平均分子量为Mn = 5456,收率为34%, 紫外光谱的最大吸收峰为342 nm。
实施例18
在一个装有磁力搅拌器,恒压滴液漏斗、回流冷凝管、以及导气口装置的50 mL三口圆底烧瓶中放置(0.6 g, 4 mmol) 金属钐粉,(0.28 g, 1 mmol) 1,2-二碘乙烷,抽真空充氮气反复进行3次。然后注入30 mL新蒸馏的四氢呋喃溶剂,在氮气保护下,室温搅拌1小时,形成蓝黑色的SmI2。加入(0.5 mmol)苯基乙基二氯硅烷,加热回流6小时后,冷却至室温,加入(0.5 mmol)R-联萘酚,结构式如手性醇(f)所示,搅拌1小时。反应结束后,用乙醚萃取(3×20 mL),有机层用饱和碳酸氢钠溶液洗涤二次(2×10 mL), 饱和食盐水溶液洗涤三次(3×10 mL)。有机层用无水硫酸镁干燥,过滤,蒸去溶剂后,用无水四氢呋喃(2 mL)溶解后,用无水乙醇再沉淀,得到光学活性的聚苯基丙基硅烷。
聚苯基丙基硅烷平均分子量分布为Mw/Mn = 1.52,平均分子量为Mn = 6756,收率为34%。紫外光谱的最大吸收峰为342 nm。
